| [1] |
Preskill J. Quantum Computing in the NISQ era and beyond. Quantum, 2018, 2: 79. doi: 10.22331/q-2018-08-06-79
|
| [2] |
McArdle S, Endo S, Aspuru-Guzik A, et al. Quantum computational chemistry. Rev. Mod. Phys., 2020, 92: 015003. doi: 10.1103/RevModPhys.92.015003
|
| [3] |
Yung M H, Casanova J, Mezzacapo A, et al. From transistor to trapped-ion computers for quantum chemistry. Sci. Rep., 2014, 4 (1): 3589. doi: 10.1038/srep03589
|
| [4] |
Tilly J, Chen H, Cao S, et al. The Variational Quantum Eigensolver: a review of methods and best practices. 2021. https://arxiv.org/abs/2111.05176. Accessed August 1, 2022.
|
| [5] |
Cerezo M, Arrasmith A, Babbush R, et al. Variational quantum algorithms. Nat. Rev. Phys., 2021, 3 (9): 625–644. doi: 10.1038/s42254-021-00348-9
|
| [6] |
Magann A B, Arenz C, Grace M D, et al. From pulses to circuits and back again: A quantum optimal control perspective on variational quantum algorithms. PRX Quantum, 2021, 2: 010101. doi: 10.1103/PRXQuantum.2.010101
|
| [7] |
Fedorov D A, Peng B, Govind N, et al. VQE method: a short survey and recent developments. Mater. Theory, 2022, 6: 2. doi: 10.1186/s41313-021-00032-6
|
| [8] |
Cao Y, Romero J, Olson J P, et al. Quantum chemistry in the age of quantum computing. Chem. Rev., 2019, 119: 10856–10915. doi: 10.1021/acs.chemrev.8b00803
|
| [9] |
Aspuru-Guzik A, Dutoi A D, Love P J, et al. Simulated quantum computation of molecular energies. Science, 2005, 309: 1704–1707. doi: 10.1126/science.1113479
|
| [10] |
Peruzzo A, McClean J, Shadbolt P, et al. A variational eigenvalue solver on a photonic quantum processor. Nat. Commun., 2014, 5: 4213. doi: 10.1038/ncomms5213
|
| [11] |
Hempel C, Maier C, Romero J, et al. Quantum chemistry calculations on a trapped-ion quantum simulator. Phys. Rev. X, 2018, 8: 031022. doi: 10.1103/PhysRevX.8.031022
|
| [12] |
Nam Y, Chen J S, Pisenti N C, et al. Ground-state energy estimation of the water molecule on a trapped-ion quantum computer. npj Quantum Inf., 2020, 6: 33. doi: 10.1038/s41534-020-0259-3
|
| [13] |
O’Malley P J J, Babbush R, Kivlichan I D, et al. Scalable quantum simulation of molecular energies. Phys. Rev. X, 2016, 6: 031007. doi: 10.1103/PhysRevX.6.031007
|
| [14] |
Kandala A, Mezzacapo A, Temme K, et al. Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets. Nature, 2017, 549 (7671): 242–246. doi: 10.1038/nature23879
|
| [15] |
Colless J I, Ramasesh V V, Dahlen D, et al. Computation of molecular spectra on a quantum processor with an error-resilient algorithm. Phys. Rev. X, 2018, 8: 011021. doi: 10.1103/PhysRevX.8.011021
|
| [16] |
McClean J R, Romero J, Babbush R, et al. The theory of variational hybrid quantum-classical algorithms. New J. Phys., 2016, 18: 023023. doi: 10.1088/1367-2630/18/2/023023
|
| [17] |
Lanyon B P, Whitfield J D, Gillett G G, et al. Towards quantum chemistry on a quantum computer. Nat. Chem., 2010, 2: 106–111. doi: 10.1038/nchem.483
|
| [18] |
Higgott O, Wang D, Brierley S. Variational quantum computation of excited states. Quantum, 2019, 3: 156. doi: 10.22331/q-2019-07-01-156
|
| [19] |
McClean J R, Kimchi-Schwartz M E, Carter J, et al. Hybrid quantum-classical hierarchy for mitigation of decoherence and determination of excited states. Phys. Rev. A, 2017, 95: 042308. doi: 10.1103/PhysRevA.95.042308
|
| [20] |
Liu J, Fan Y, Li Z, et al. Quantum algorithms for electronic structures: basis sets and boundary conditions. Chem. Soc. Rev., 2022, 51: 3263–3279. doi: 10.1039/D1CS01184G
|
| [21] |
McClean J R, Boixo S, Smelyanskiy V N, et al. Barren plateaus in quantum neural network training landscapes. Nat. Commun., 2018, 9 (1): 4812. doi: 10.1038/s41467-018-07090-4
|
| [22] |
Napp J. Quantifying the barren plateau phenomenon for a model of unstructured variational ansätze. 2022. https://arxiv.org/abs/2203.06174. Accessed August 1, 2022.
|
| [23] |
Anschuetz E R, Kiani B T. Beyond barren plateaus: Quantum variational algorithms are swamped with traps. 2022. https://arxiv.org/abs/2205.05786. Accessed August 1, 2022.
|
| [24] |
Arute F, Arya K, Babbush R, et al. Hartree-Fock on a superconducting qubit quantum computer. Science, 2020, 369 (6507): 1084–1089. doi: 10.1126/science.abb9811
|
| [25] |
Huggins W J, O’Gorman B A, Rubin N C, et al. Unbiasing fermionic quantum Monte Carlo with a quantum computer. Nature, 2022, 603 (7901): 416–420. doi: 10.1038/s41586-021-04351-z
|
| [26] |
Bartlett R J, Kucharski S A, Noga J. Alternative coupled-cluster ansätze Ⅱ. The unitary coupled-cluster method. Chem. Phys. Lett., 1989, 155: 133–140. doi: 10.1016/S0009-2614(89)87372-5
|
| [27] |
Taube A G, Bartlett R J. New perspectives on unitary coupled-cluster theory. Int. J. Quantum Chem., 2006, 106: 3393–3401. doi: 10.1002/qua.21198
|
| [28] |
Steiger D S, Häner T, Troyer M. ProjectQ: an open source software framework for quantum computing. Quantum, 2018, 2: 49. doi: 10.22331/q-2018-01-31-49
|
| [29] |
ANIS M S, Mitchell A, Abraham H, et al. Qiskit. 2021. https://github.com/Qiskit/qiskit. Accessed August 1, 2022.
|
| [30] |
Suzuki Y, Kawase Y, Masumura Y, et al. Qulacs: a fast and versatile quantum circuit simulator for research purpose. Quantum, 2021, 5: 559. doi: 10.22331/q-2021-10-06-559
|
| [31] |
Luo X Z, Liu J G, Zhang P, et al. Yao.jl: Extensible, efficient framework for quantum algorithm design. Quantum, 2020, 4: 341. doi: 10.22331/q-2020-10-11-341
|
| [32] |
Bergholm V, Izaac J, Schuld M, et al. PennyLane: Automatic differentiation of hybrid quantum-classical computations. 2018. https://arxiv.org/abs/1811.04968. Accessed August 1, 2022.
|
| [33] |
Cao C, Hu J, Zhang W, et al. Progress toward larger molecular simulation on a quantum computer: Simulating a system with up to 28 qubits accelerated by point-group symmetry. Phys. Rev. A, 2022, 105: 062452. doi: 10.1103/PhysRevA.105.062452
|
| [34] |
Bezanson J, Edelman A, Karpinski S, et al. Julia: A fresh approach to numerical computing. SIAM Review, 2017, 59 (1): 65–98. doi: 10.1137/141000671
|
| [35] |
Sun Q, Berkelbach T C, Blunt N S, et al. PySCF: the Python-based simulations of chemistry framework. WIREs Comput. Mol. Sci., 2018, 8: e1340. doi: 10.1002/wcms.1340
|
| [36] |
Orús R. A practical introduction to tensor networks: Matrix product states and projected entangled pair states. Ann. Phys., 2014, 349: 117–158. doi: 10.1016/j.aop.2014.06.013
|
| [37] |
Schollwöck U. The density-matrix renormalization group in the age of matrix product states. Ann. Phys., 2011, 326 (1): 96–192. doi: 10.1016/j.aop.2010.09.012
|
| [38] |
Harris C R, Millman K J, van der Walt S J, et al. Array programming with NumPy. Nature, 2020, 585 (7825): 357–362. doi: 10.1038/s41586-020-2649-2
|
| [39] |
Virtanen P, Gommers R, Oliphant T E, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods, 2020, 17: 261–272. doi: 10.1038/s41592-019-0686-2
|
| [40] |
Paszke A, Gross S, Massa F, et al. PyTorch: An imperative style, high-performance deep learning library. In: Wallach H, Larochelle H, Beygelzimer A, editors. Advances in Neural Information Processing Systems 32. New York: Curran Associates, Inc., 2019: 8024–8035.
|
| [41] |
Jordan P, Wigner E. Über das paulische äquivalenzverbot. Z. Physik, 1928, 47: 631–651. doi: 10.1007/BF01331938
|
| [42] |
Seeley J T, Richard M J, Love P J. The Bravyi-Kitaev transformation for quantum computation of electronic structure. J. Chem. Phys., 2012, 137: 224109. doi: 10.1063/1.4768229
|
| [43] |
Tranter A, Love P J, Mintert F, et al. A comparison of the Bravyi-Kitaev and Jordan-Wigner transformations for the quantum simulation of quantum chemistry. J. Chem. Theory Comput., 2018, 14: 5617–5630. doi: 10.1021/acs.jctc.8b00450
|
| [44] |
Liu J, Wan L Y, Li Z Y, et al. Simulating periodic systems on a quantum computer using molecular orbitals. J. Chem. Theory Comput., 2020, 16: 6904–6914. doi: 10.1021/acs.jctc.0c00881
|
| [45] |
Fan Y, Liu J, Li Z Y, et al. Equation-of-motion theory to calculate accurate band structures with a quantum computer. J. Phys. Chem. Lett., 2021, 12 (36): 8833–8840. doi: 10.1021/acs.jpclett.1c02153
|
| [46] |
Smith D G A, Gray J. opt_einsum - A Python package for optimizing contraction order for einsum-like expressions. J. Open Source Softw., 2018, 3 (26): 753. doi: 10.21105/joss.00753
|
| [47] |
McClean J R, Sung K J, Kivlichan I D, et al. OpenFermion: The electronic structure package for quantum computers. 2017. https://arxiv.org/abs/1710.07629. Accessed August 1, 2022.
|
| [48] |
Liu J G, Zhang Y H, Wan Y, et al. Variational quantum eigensolver with fewer qubits. Phys. Rev. Res., 2019, 1: 023025. doi: 10.1103/PhysRevResearch.1.023025
|
| [49] |
Haghshenas R, Gray J, Potter A C, et al. Variational power of quantum circuit tensor networks. Phys. Rev. X, 2022, 12: 011047. doi: 10.1103/PhysRevX.12.011047
|
| [50] |
Nguyen D, Mikushin D, Man-Hong Y. HiQ-ProjectQ: Towards user-friendly and high-performance quantum computing on GPUs. In: 2021 Design, Automation & Test in Europe Conference & Exhibition (DATE). IEEE, 2021: 1056–1061.
|
| [51] |
MindQuantum Developer. MindQuantum, version 0.6.0. 2021. https://gitee.com/mindspore/mindquantum. Accessed August 1, 2022.
|
| [52] |
Cirq Developers. Cirq. 2022. https://github.com/quantumlib/Cirq. Accessed August 1, 2022.
|
| [53] |
Paddle Quantum Developers. Paddle Quantum. 2020. https://github.com/PaddlePaddle/Quantum. Accessed August 1, 2022.
|
| [54] |
Jones T, Brown A, Bush I, et al. QuEST and high performance simulation of quantum computers. Sci. Rep., 2019, 9 (1): 10736. doi: 10.1038/s41598-019-47174-9
|
| [55] |
Kottmann J S, Alperin-Lea S, Tamayo-Mendoza T, et al. TEQUILA: a platform for rapid development of quantum algorithms. Quantum Sci. Technol., 2021, 6 (2): 024009. doi: 10.1088/2058-9565/abe567
|
| [56] |
Stair N H, Evangelista F A. QForte: An efficient state-vector emulator and quantum algorithms library for molecular electronic structure. J. Chem. Theory Comput., 2022, 18 (3): 1555–1568. doi: 10.1021/acs.jctc.1c01155
|
| [57] |
McCaskey A J, Lyakh D I, Dumitrescu E F, et al. XACC: a system-level software infrastructure for heterogeneous quantum-classical computing. Quantum Sci. Technol., 2020, 5 (2): 024002. doi: 10.1088/2058-9565/ab6bf6
|
| [58] |
Guo C, Liu Y, Xiong M, et al. General-purpose quantum circuit simulator with projected entangled-pair states and the quantum supremacy frontier. Phys. Rev. Lett., 2019, 123: 190501. doi: 10.1103/PhysRevLett.123.190501
|
| [59] |
Guo C, Zhao Y, Huang H L. Verifying random quantum circuits with arbitrary geometry using tensor network states algorithm. Phys. Rev. Lett., 2021, 126: 070502. doi: 10.1103/PhysRevLett.126.070502
|
| [60] |
Liu X, Guo C, Liu Y, et al. Redefining the quantum supremacy baseline with a new generation sunway supercomputer. 2021. https://arxiv.org/abs/2111.01066. Accessed August 1, 2022.
|
| [61] |
McCaskey A, Dumitrescu E, Chen M, et al. Validating quantum-classical programming models with tensor network simulations. PLoS ONE, 2018, 13 (12): e0206704. doi: 10.1371/journal.pone.0206704
|
| [62] |
Pfeifer R N C, Haegeman J, Verstraete F. Faster identification of optimal contraction sequences for tensor networks. Phys. Rev. E, 2014, 90: 033315. doi: 10.1103/PhysRevE.90.033315
|
| [63] |
Vidal G. Efficient classical simulation of slightly entangled quantum computations. Phys. Rev. Lett., 2003, 91: 147902. doi: 10.1103/PhysRevLett.91.147902
|
| [64] |
Vidal G. Classical simulation of infinite-size quantum lattice systems in one spatial dimension. Phys. Rev. Lett., 2007, 98: 070201. doi: 10.1103/PhysRevLett.98.070201
|
| [65] |
Guo C. QuantumSpins. 2020. https://github.com/guochu/QuantumSpins. Accessed May 17, 2022.
|
| [66] |
Gomez A N, Ren M, Urtasun R, et al. The reversible residual network: Backpropagation without storing activations. 2017. https://arxiv.org/abs/1707.04585. Accessed October 21, 2022.
|
| [67] |
Chen R T Q, Rubanova Y, Bettencourt J, et al. Neural ordinary differential equations. 2019. https://arxiv.org/abs/1806.07366. Accessed October 21, 2022.
|
| [68] |
Jones T, Gacon J. Efficient calculation of gradients in classical simulations of variational quantum algorithms. 2020. https://arxiv.org/abs/2009.02823. Accessed August 1, 2022.
|
| [69] |
Bulik I W, Henderson T M, Scuseria G E. Can single-reference coupled cluster theory describe static correlation? J. Chem. Theory Comput., 2015, 11 (7): 3171–3179. doi: 10.1021/acs.jctc.5b00422
|
| [70] |
Grimsley H R, Claudino D, Economou S E, et al. Is the Trotterized UCCSD ansatz chemically well-defined? J. Chem. Theory Comput., 2020, 16: 1–6. doi: 10.1021/acs.jctc.9b01083
|
| [71] |
Babbush R, McClean J, Wecker D, et al. Chemical basis of Trotter-Suzuki errors in quantum chemistry simulation. Phys. Rev. A, 2015, 91: 022311. doi: 10.1103/PhysRevA.91.022311
|
| [72] |
Bravyi S, Gambetta J M, Mezzacapo A, et al. Tapering off qubits to simulate fermionic Hamiltonians. 2017. https://arxiv.org/abs/1701.08213. Accessed August 1, 2022.
|
| [73] |
Yordanov Y S, Armaos V, Barnes C H W, et al. Qubit-excitation-based adaptive variational quantum eigensolver. Commun. Phys., 2021, 4 (1): 228. doi: 10.1038/s42005-021-00730-0
|
| [74] |
Ryabinkin I G, Yen T C, Genin S N, et al. Qubit coupled cluster method: A systematic approach to quantum chemistry on a quantum computer. J. Chem. Theory Comput., 2018, 14 (12): 6317–6326. doi: 10.1021/acs.jctc.8b00932
|
| [75] |
Ryabinkin I G, Lang R A, Genin S N, et al. Iterative qubit coupled cluster approach with efficient screening of generators. J. Chem. Theory Comput., 2020, 16 (2): 1055–1063. doi: 10.1021/acs.jctc.9b01084
|
| [76] |
Grimsley H R, Economou S E, Barnes E, et al. An adaptive variational algorithm for exact molecular simulations on a quantum computer. Nat. Commun., 2019, 10: 3007. doi: 10.1038/s41467-019-10988-2
|
| [77] |
Krylov A I. Equation-of-motion coupled-cluster methods for open-shell and electronically excited species: The hitchhiker’s guide to Fock space. Annu. Rev. Phys. Chem., 2008, 59: 433–462. doi: 10.1146/annurev.physchem.59.032607.093602
|
| [78] |
Ollitrault P J, Kandala A, Chen C F, et al. Quantum equation of motion for computing molecular excitation energies on a noisy quantum processor. Phys. Rev. Res., 2020, 2: 043140. doi: 10.1103/PhysRevResearch.2.043140
|
| [79] |
Benedikt U, Auer A A, Jensen F. Optimization of augmentation functions for correlated calculations of spin-spin coupling constants and related properties. J. Chem. Phys., 2008, 129 (6): 064111. doi: 10.1063/1.2962973
|
 [For_publication]Supporting_Information_FULL.pdf
[For_publication]Supporting_Information_FULL.pdf
|

|
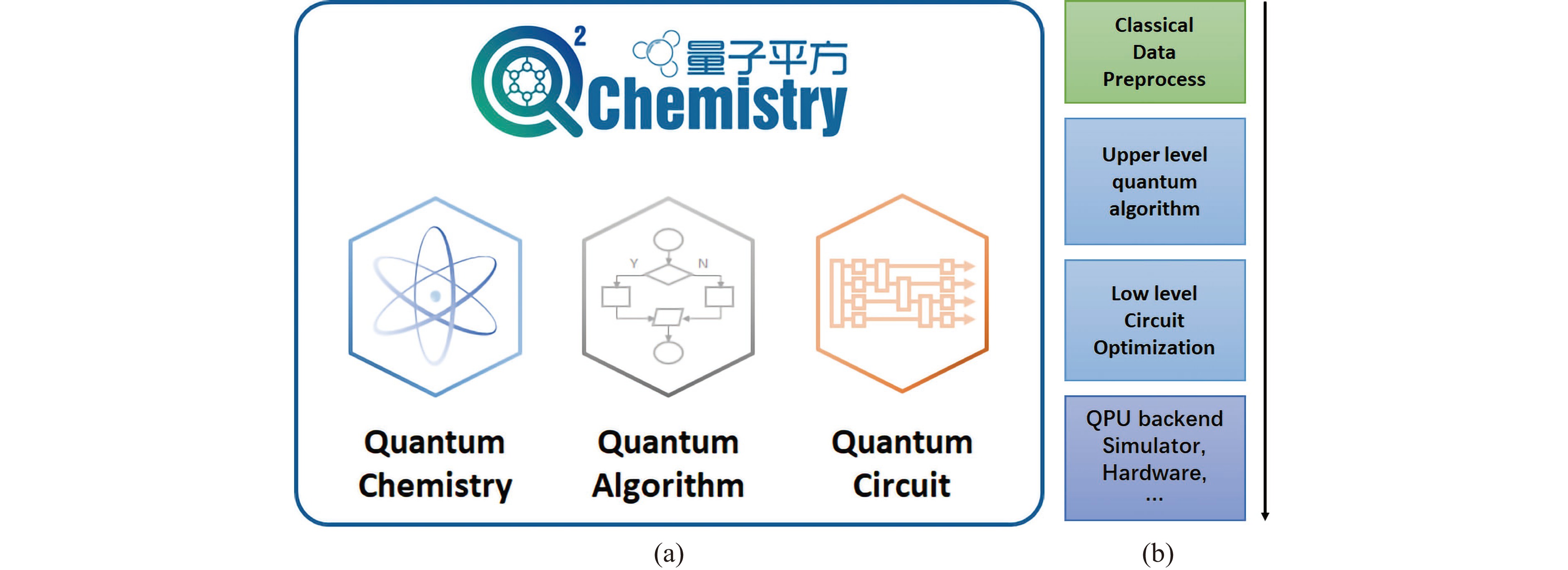
Figure 1. (a) The framework of Q2Chemistry. (b) A typical workflow of solving a chemical problem using a quantum algorithm.
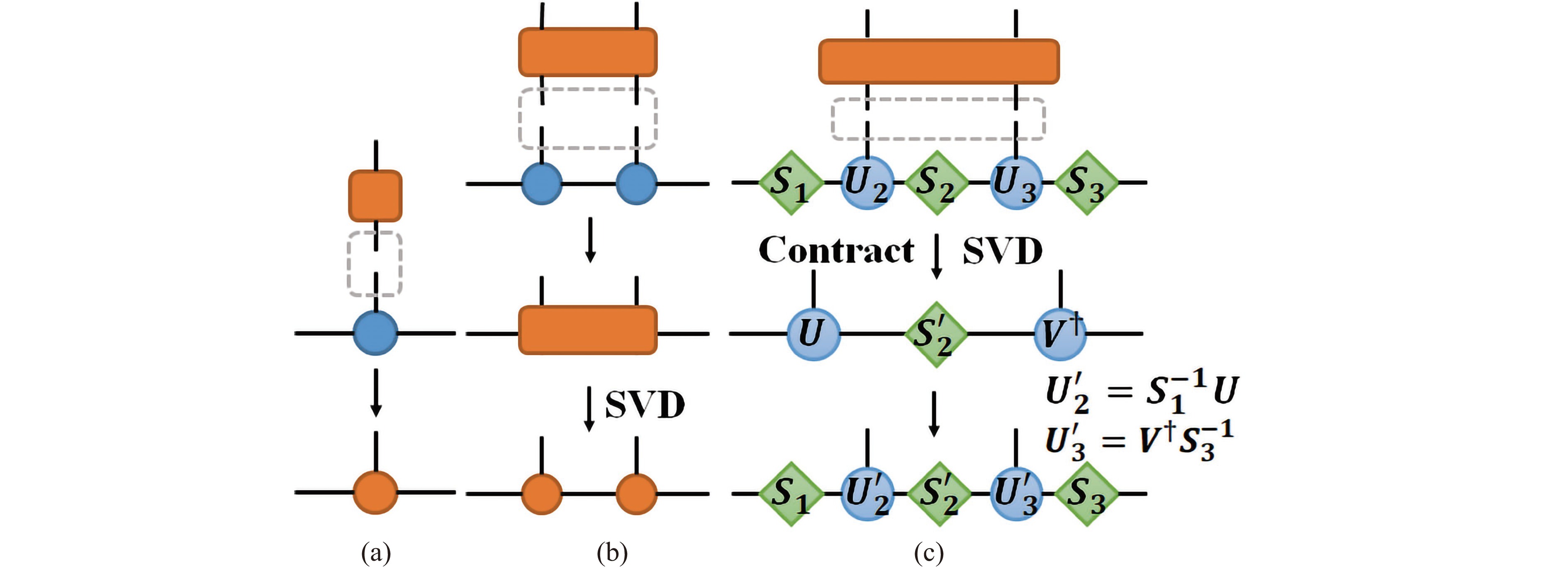
Figure 2. (a) Applying a single qubit gate on the MPS quantum state is simulated by simply a local contraction. (b) Applying a two-qubit gate on neighboring qubits generally has 2 steps: (i) reshape the two-qubit gate into a 4-dimensional tensor and contract with the qubits to form a two-qubit tensor; (ii) perform a singular value decomposition to restore the two-qubit tensor back to the MPS formulation. Postprocessing is usually required to maintain normalization or canonicalization of MPS tensors. (c) Auxiliary matrices that contain truncated and normalized singular values are used to normalize the quantum state.
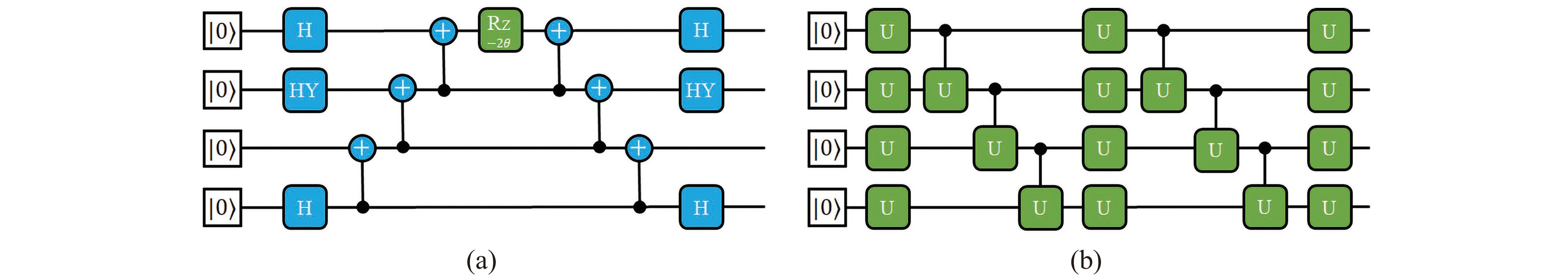
Figure
3.
(a) The quantum circuit corresponding to the operator
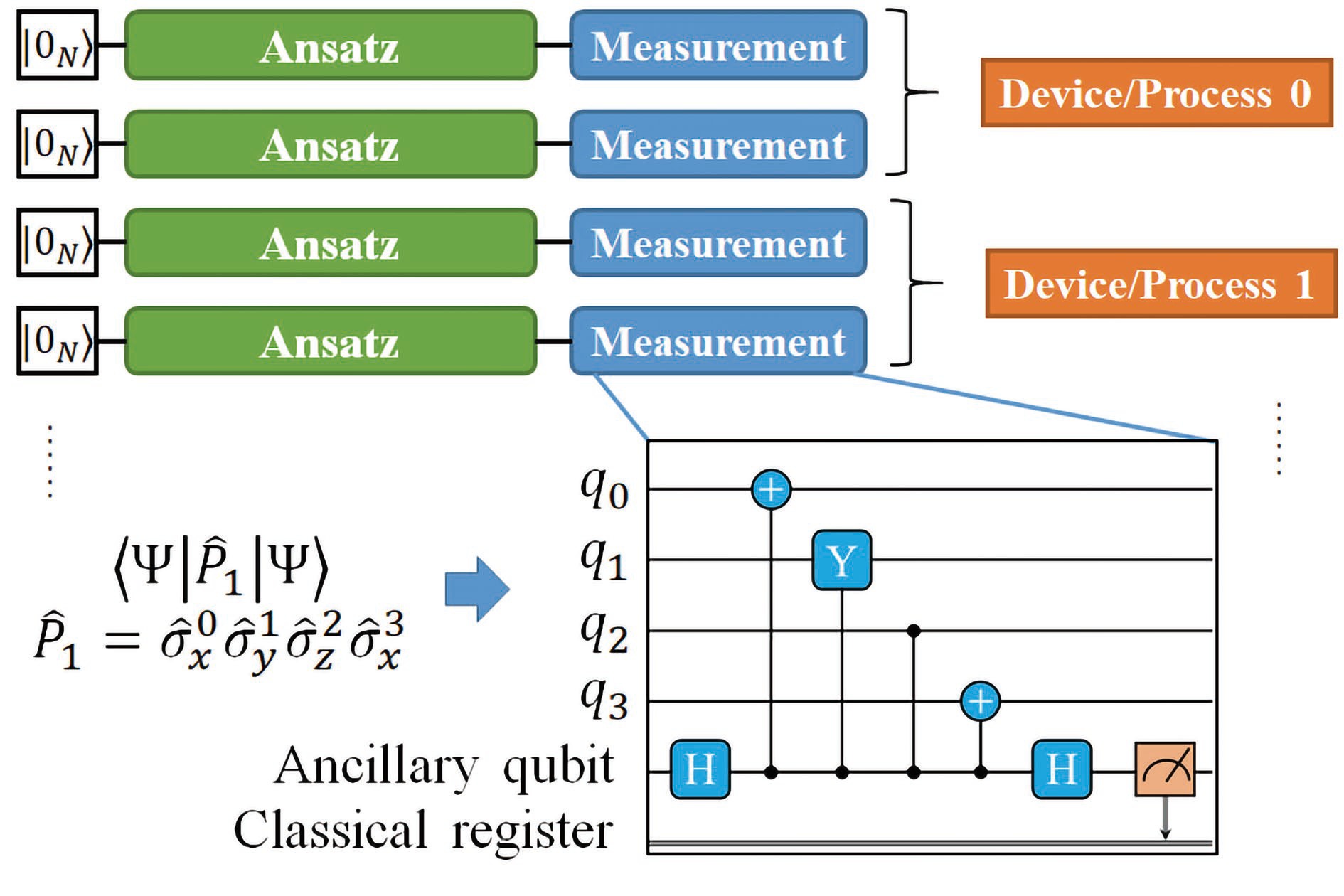
Figure
4.
Evaluating the expectation value of a linear combination of Pauli strings using multiple quantum devices or simulator processes.
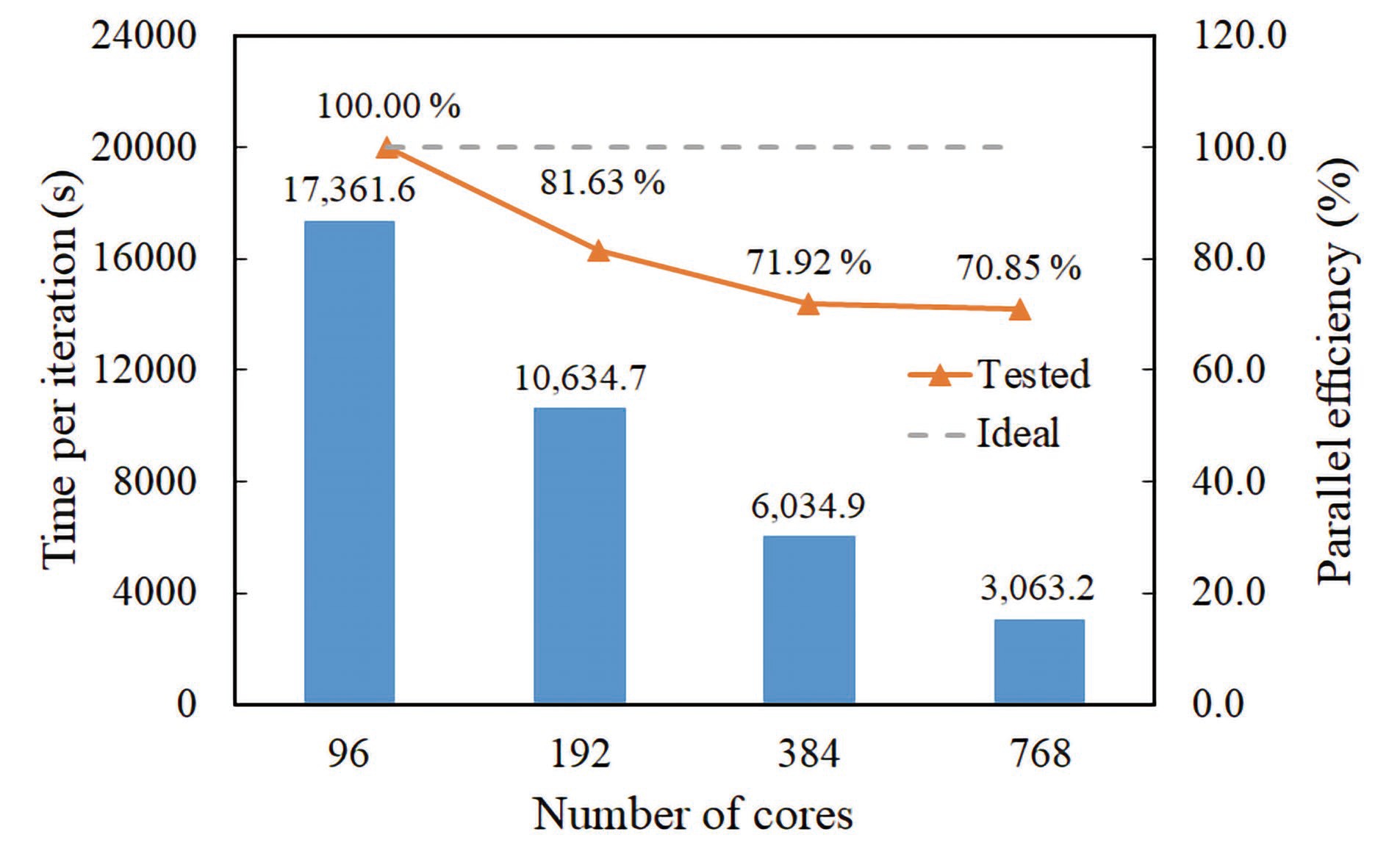
Figure
5.
Simulating the Cr
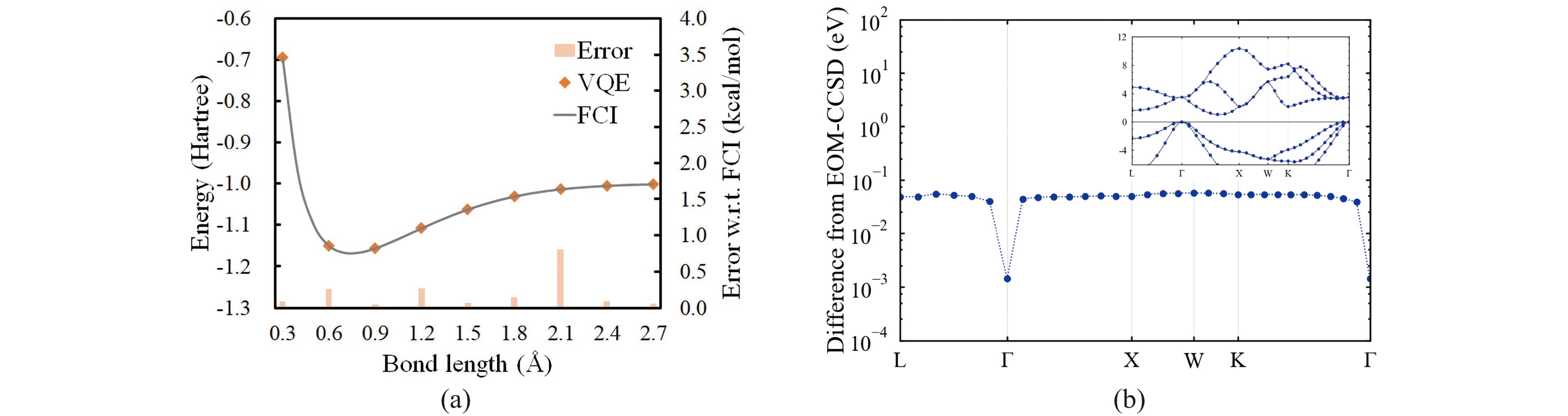
Figure
6.
(a) The VQE optimized potential energy curve of H

Figure . Code Example 1. Interface to quantum devices in the qcircuit module.
| [1] |
Preskill J. Quantum Computing in the NISQ era and beyond. Quantum, 2018, 2: 79. doi: 10.22331/q-2018-08-06-79
|
| [2] |
McArdle S, Endo S, Aspuru-Guzik A, et al. Quantum computational chemistry. Rev. Mod. Phys., 2020, 92: 015003. doi: 10.1103/RevModPhys.92.015003
|
| [3] |
Yung M H, Casanova J, Mezzacapo A, et al. From transistor to trapped-ion computers for quantum chemistry. Sci. Rep., 2014, 4 (1): 3589. doi: 10.1038/srep03589
|
| [4] |
Tilly J, Chen H, Cao S, et al. The Variational Quantum Eigensolver: a review of methods and best practices. 2021. https://arxiv.org/abs/2111.05176. Accessed August 1, 2022.
|
| [5] |
Cerezo M, Arrasmith A, Babbush R, et al. Variational quantum algorithms. Nat. Rev. Phys., 2021, 3 (9): 625–644. doi: 10.1038/s42254-021-00348-9
|
| [6] |
Magann A B, Arenz C, Grace M D, et al. From pulses to circuits and back again: A quantum optimal control perspective on variational quantum algorithms. PRX Quantum, 2021, 2: 010101. doi: 10.1103/PRXQuantum.2.010101
|
| [7] |
Fedorov D A, Peng B, Govind N, et al. VQE method: a short survey and recent developments. Mater. Theory, 2022, 6: 2. doi: 10.1186/s41313-021-00032-6
|
| [8] |
Cao Y, Romero J, Olson J P, et al. Quantum chemistry in the age of quantum computing. Chem. Rev., 2019, 119: 10856–10915. doi: 10.1021/acs.chemrev.8b00803
|
| [9] |
Aspuru-Guzik A, Dutoi A D, Love P J, et al. Simulated quantum computation of molecular energies. Science, 2005, 309: 1704–1707. doi: 10.1126/science.1113479
|
| [10] |
Peruzzo A, McClean J, Shadbolt P, et al. A variational eigenvalue solver on a photonic quantum processor. Nat. Commun., 2014, 5: 4213. doi: 10.1038/ncomms5213
|
| [11] |
Hempel C, Maier C, Romero J, et al. Quantum chemistry calculations on a trapped-ion quantum simulator. Phys. Rev. X, 2018, 8: 031022. doi: 10.1103/PhysRevX.8.031022
|
| [12] |
Nam Y, Chen J S, Pisenti N C, et al. Ground-state energy estimation of the water molecule on a trapped-ion quantum computer. npj Quantum Inf., 2020, 6: 33. doi: 10.1038/s41534-020-0259-3
|
| [13] |
O’Malley P J J, Babbush R, Kivlichan I D, et al. Scalable quantum simulation of molecular energies. Phys. Rev. X, 2016, 6: 031007. doi: 10.1103/PhysRevX.6.031007
|
| [14] |
Kandala A, Mezzacapo A, Temme K, et al. Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets. Nature, 2017, 549 (7671): 242–246. doi: 10.1038/nature23879
|
| [15] |
Colless J I, Ramasesh V V, Dahlen D, et al. Computation of molecular spectra on a quantum processor with an error-resilient algorithm. Phys. Rev. X, 2018, 8: 011021. doi: 10.1103/PhysRevX.8.011021
|
| [16] |
McClean J R, Romero J, Babbush R, et al. The theory of variational hybrid quantum-classical algorithms. New J. Phys., 2016, 18: 023023. doi: 10.1088/1367-2630/18/2/023023
|
| [17] |
Lanyon B P, Whitfield J D, Gillett G G, et al. Towards quantum chemistry on a quantum computer. Nat. Chem., 2010, 2: 106–111. doi: 10.1038/nchem.483
|
| [18] |
Higgott O, Wang D, Brierley S. Variational quantum computation of excited states. Quantum, 2019, 3: 156. doi: 10.22331/q-2019-07-01-156
|
| [19] |
McClean J R, Kimchi-Schwartz M E, Carter J, et al. Hybrid quantum-classical hierarchy for mitigation of decoherence and determination of excited states. Phys. Rev. A, 2017, 95: 042308. doi: 10.1103/PhysRevA.95.042308
|
| [20] |
Liu J, Fan Y, Li Z, et al. Quantum algorithms for electronic structures: basis sets and boundary conditions. Chem. Soc. Rev., 2022, 51: 3263–3279. doi: 10.1039/D1CS01184G
|
| [21] |
McClean J R, Boixo S, Smelyanskiy V N, et al. Barren plateaus in quantum neural network training landscapes. Nat. Commun., 2018, 9 (1): 4812. doi: 10.1038/s41467-018-07090-4
|
| [22] |
Napp J. Quantifying the barren plateau phenomenon for a model of unstructured variational ansätze. 2022. https://arxiv.org/abs/2203.06174. Accessed August 1, 2022.
|
| [23] |
Anschuetz E R, Kiani B T. Beyond barren plateaus: Quantum variational algorithms are swamped with traps. 2022. https://arxiv.org/abs/2205.05786. Accessed August 1, 2022.
|
| [24] |
Arute F, Arya K, Babbush R, et al. Hartree-Fock on a superconducting qubit quantum computer. Science, 2020, 369 (6507): 1084–1089. doi: 10.1126/science.abb9811
|
| [25] |
Huggins W J, O’Gorman B A, Rubin N C, et al. Unbiasing fermionic quantum Monte Carlo with a quantum computer. Nature, 2022, 603 (7901): 416–420. doi: 10.1038/s41586-021-04351-z
|
| [26] |
Bartlett R J, Kucharski S A, Noga J. Alternative coupled-cluster ansätze Ⅱ. The unitary coupled-cluster method. Chem. Phys. Lett., 1989, 155: 133–140. doi: 10.1016/S0009-2614(89)87372-5
|
| [27] |
Taube A G, Bartlett R J. New perspectives on unitary coupled-cluster theory. Int. J. Quantum Chem., 2006, 106: 3393–3401. doi: 10.1002/qua.21198
|
| [28] |
Steiger D S, Häner T, Troyer M. ProjectQ: an open source software framework for quantum computing. Quantum, 2018, 2: 49. doi: 10.22331/q-2018-01-31-49
|
| [29] |
ANIS M S, Mitchell A, Abraham H, et al. Qiskit. 2021. https://github.com/Qiskit/qiskit. Accessed August 1, 2022.
|
| [30] |
Suzuki Y, Kawase Y, Masumura Y, et al. Qulacs: a fast and versatile quantum circuit simulator for research purpose. Quantum, 2021, 5: 559. doi: 10.22331/q-2021-10-06-559
|
| [31] |
Luo X Z, Liu J G, Zhang P, et al. Yao.jl: Extensible, efficient framework for quantum algorithm design. Quantum, 2020, 4: 341. doi: 10.22331/q-2020-10-11-341
|
| [32] |
Bergholm V, Izaac J, Schuld M, et al. PennyLane: Automatic differentiation of hybrid quantum-classical computations. 2018. https://arxiv.org/abs/1811.04968. Accessed August 1, 2022.
|
| [33] |
Cao C, Hu J, Zhang W, et al. Progress toward larger molecular simulation on a quantum computer: Simulating a system with up to 28 qubits accelerated by point-group symmetry. Phys. Rev. A, 2022, 105: 062452. doi: 10.1103/PhysRevA.105.062452
|
| [34] |
Bezanson J, Edelman A, Karpinski S, et al. Julia: A fresh approach to numerical computing. SIAM Review, 2017, 59 (1): 65–98. doi: 10.1137/141000671
|
| [35] |
Sun Q, Berkelbach T C, Blunt N S, et al. PySCF: the Python-based simulations of chemistry framework. WIREs Comput. Mol. Sci., 2018, 8: e1340. doi: 10.1002/wcms.1340
|
| [36] |
Orús R. A practical introduction to tensor networks: Matrix product states and projected entangled pair states. Ann. Phys., 2014, 349: 117–158. doi: 10.1016/j.aop.2014.06.013
|
| [37] |
Schollwöck U. The density-matrix renormalization group in the age of matrix product states. Ann. Phys., 2011, 326 (1): 96–192. doi: 10.1016/j.aop.2010.09.012
|
| [38] |
Harris C R, Millman K J, van der Walt S J, et al. Array programming with NumPy. Nature, 2020, 585 (7825): 357–362. doi: 10.1038/s41586-020-2649-2
|
| [39] |
Virtanen P, Gommers R, Oliphant T E, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods, 2020, 17: 261–272. doi: 10.1038/s41592-019-0686-2
|
| [40] |
Paszke A, Gross S, Massa F, et al. PyTorch: An imperative style, high-performance deep learning library. In: Wallach H, Larochelle H, Beygelzimer A, editors. Advances in Neural Information Processing Systems 32. New York: Curran Associates, Inc., 2019: 8024–8035.
|
| [41] |
Jordan P, Wigner E. Über das paulische äquivalenzverbot. Z. Physik, 1928, 47: 631–651. doi: 10.1007/BF01331938
|
| [42] |
Seeley J T, Richard M J, Love P J. The Bravyi-Kitaev transformation for quantum computation of electronic structure. J. Chem. Phys., 2012, 137: 224109. doi: 10.1063/1.4768229
|
| [43] |
Tranter A, Love P J, Mintert F, et al. A comparison of the Bravyi-Kitaev and Jordan-Wigner transformations for the quantum simulation of quantum chemistry. J. Chem. Theory Comput., 2018, 14: 5617–5630. doi: 10.1021/acs.jctc.8b00450
|
| [44] |
Liu J, Wan L Y, Li Z Y, et al. Simulating periodic systems on a quantum computer using molecular orbitals. J. Chem. Theory Comput., 2020, 16: 6904–6914. doi: 10.1021/acs.jctc.0c00881
|
| [45] |
Fan Y, Liu J, Li Z Y, et al. Equation-of-motion theory to calculate accurate band structures with a quantum computer. J. Phys. Chem. Lett., 2021, 12 (36): 8833–8840. doi: 10.1021/acs.jpclett.1c02153
|
| [46] |
Smith D G A, Gray J. opt_einsum - A Python package for optimizing contraction order for einsum-like expressions. J. Open Source Softw., 2018, 3 (26): 753. doi: 10.21105/joss.00753
|
| [47] |
McClean J R, Sung K J, Kivlichan I D, et al. OpenFermion: The electronic structure package for quantum computers. 2017. https://arxiv.org/abs/1710.07629. Accessed August 1, 2022.
|
| [48] |
Liu J G, Zhang Y H, Wan Y, et al. Variational quantum eigensolver with fewer qubits. Phys. Rev. Res., 2019, 1: 023025. doi: 10.1103/PhysRevResearch.1.023025
|
| [49] |
Haghshenas R, Gray J, Potter A C, et al. Variational power of quantum circuit tensor networks. Phys. Rev. X, 2022, 12: 011047. doi: 10.1103/PhysRevX.12.011047
|
| [50] |
Nguyen D, Mikushin D, Man-Hong Y. HiQ-ProjectQ: Towards user-friendly and high-performance quantum computing on GPUs. In: 2021 Design, Automation & Test in Europe Conference & Exhibition (DATE). IEEE, 2021: 1056–1061.
|
| [51] |
MindQuantum Developer. MindQuantum, version 0.6.0. 2021. https://gitee.com/mindspore/mindquantum. Accessed August 1, 2022.
|
| [52] |
Cirq Developers. Cirq. 2022. https://github.com/quantumlib/Cirq. Accessed August 1, 2022.
|
| [53] |
Paddle Quantum Developers. Paddle Quantum. 2020. https://github.com/PaddlePaddle/Quantum. Accessed August 1, 2022.
|
| [54] |
Jones T, Brown A, Bush I, et al. QuEST and high performance simulation of quantum computers. Sci. Rep., 2019, 9 (1): 10736. doi: 10.1038/s41598-019-47174-9
|
| [55] |
Kottmann J S, Alperin-Lea S, Tamayo-Mendoza T, et al. TEQUILA: a platform for rapid development of quantum algorithms. Quantum Sci. Technol., 2021, 6 (2): 024009. doi: 10.1088/2058-9565/abe567
|
| [56] |
Stair N H, Evangelista F A. QForte: An efficient state-vector emulator and quantum algorithms library for molecular electronic structure. J. Chem. Theory Comput., 2022, 18 (3): 1555–1568. doi: 10.1021/acs.jctc.1c01155
|
| [57] |
McCaskey A J, Lyakh D I, Dumitrescu E F, et al. XACC: a system-level software infrastructure for heterogeneous quantum-classical computing. Quantum Sci. Technol., 2020, 5 (2): 024002. doi: 10.1088/2058-9565/ab6bf6
|
| [58] |
Guo C, Liu Y, Xiong M, et al. General-purpose quantum circuit simulator with projected entangled-pair states and the quantum supremacy frontier. Phys. Rev. Lett., 2019, 123: 190501. doi: 10.1103/PhysRevLett.123.190501
|
| [59] |
Guo C, Zhao Y, Huang H L. Verifying random quantum circuits with arbitrary geometry using tensor network states algorithm. Phys. Rev. Lett., 2021, 126: 070502. doi: 10.1103/PhysRevLett.126.070502
|
| [60] |
Liu X, Guo C, Liu Y, et al. Redefining the quantum supremacy baseline with a new generation sunway supercomputer. 2021. https://arxiv.org/abs/2111.01066. Accessed August 1, 2022.
|
| [61] |
McCaskey A, Dumitrescu E, Chen M, et al. Validating quantum-classical programming models with tensor network simulations. PLoS ONE, 2018, 13 (12): e0206704. doi: 10.1371/journal.pone.0206704
|
| [62] |
Pfeifer R N C, Haegeman J, Verstraete F. Faster identification of optimal contraction sequences for tensor networks. Phys. Rev. E, 2014, 90: 033315. doi: 10.1103/PhysRevE.90.033315
|
| [63] |
Vidal G. Efficient classical simulation of slightly entangled quantum computations. Phys. Rev. Lett., 2003, 91: 147902. doi: 10.1103/PhysRevLett.91.147902
|
| [64] |
Vidal G. Classical simulation of infinite-size quantum lattice systems in one spatial dimension. Phys. Rev. Lett., 2007, 98: 070201. doi: 10.1103/PhysRevLett.98.070201
|
| [65] |
Guo C. QuantumSpins. 2020. https://github.com/guochu/QuantumSpins. Accessed May 17, 2022.
|
| [66] |
Gomez A N, Ren M, Urtasun R, et al. The reversible residual network: Backpropagation without storing activations. 2017. https://arxiv.org/abs/1707.04585. Accessed October 21, 2022.
|
| [67] |
Chen R T Q, Rubanova Y, Bettencourt J, et al. Neural ordinary differential equations. 2019. https://arxiv.org/abs/1806.07366. Accessed October 21, 2022.
|
| [68] |
Jones T, Gacon J. Efficient calculation of gradients in classical simulations of variational quantum algorithms. 2020. https://arxiv.org/abs/2009.02823. Accessed August 1, 2022.
|
| [69] |
Bulik I W, Henderson T M, Scuseria G E. Can single-reference coupled cluster theory describe static correlation? J. Chem. Theory Comput., 2015, 11 (7): 3171–3179. doi: 10.1021/acs.jctc.5b00422
|
| [70] |
Grimsley H R, Claudino D, Economou S E, et al. Is the Trotterized UCCSD ansatz chemically well-defined? J. Chem. Theory Comput., 2020, 16: 1–6. doi: 10.1021/acs.jctc.9b01083
|
| [71] |
Babbush R, McClean J, Wecker D, et al. Chemical basis of Trotter-Suzuki errors in quantum chemistry simulation. Phys. Rev. A, 2015, 91: 022311. doi: 10.1103/PhysRevA.91.022311
|
| [72] |
Bravyi S, Gambetta J M, Mezzacapo A, et al. Tapering off qubits to simulate fermionic Hamiltonians. 2017. https://arxiv.org/abs/1701.08213. Accessed August 1, 2022.
|
| [73] |
Yordanov Y S, Armaos V, Barnes C H W, et al. Qubit-excitation-based adaptive variational quantum eigensolver. Commun. Phys., 2021, 4 (1): 228. doi: 10.1038/s42005-021-00730-0
|
| [74] |
Ryabinkin I G, Yen T C, Genin S N, et al. Qubit coupled cluster method: A systematic approach to quantum chemistry on a quantum computer. J. Chem. Theory Comput., 2018, 14 (12): 6317–6326. doi: 10.1021/acs.jctc.8b00932
|
| [75] |
Ryabinkin I G, Lang R A, Genin S N, et al. Iterative qubit coupled cluster approach with efficient screening of generators. J. Chem. Theory Comput., 2020, 16 (2): 1055–1063. doi: 10.1021/acs.jctc.9b01084
|
| [76] |
Grimsley H R, Economou S E, Barnes E, et al. An adaptive variational algorithm for exact molecular simulations on a quantum computer. Nat. Commun., 2019, 10: 3007. doi: 10.1038/s41467-019-10988-2
|
| [77] |
Krylov A I. Equation-of-motion coupled-cluster methods for open-shell and electronically excited species: The hitchhiker’s guide to Fock space. Annu. Rev. Phys. Chem., 2008, 59: 433–462. doi: 10.1146/annurev.physchem.59.032607.093602
|
| [78] |
Ollitrault P J, Kandala A, Chen C F, et al. Quantum equation of motion for computing molecular excitation energies on a noisy quantum processor. Phys. Rev. Res., 2020, 2: 043140. doi: 10.1103/PhysRevResearch.2.043140
|
| [79] |
Benedikt U, Auer A A, Jensen F. Optimization of augmentation functions for correlated calculations of spin-spin coupling constants and related properties. J. Chem. Phys., 2008, 129 (6): 064111. doi: 10.1063/1.2962973
|

ISSN 0253-2778
CN 34-1054/N
Copyright © Editorial Office of JUSTC, All Rights Reserved. 皖ICP备05002528号
Supported by:
Beijing Renhe Information Technology Co. Ltd












 DownLoad:
DownLoad: